包装系统是指容纳和保护药品的所有包装组件,包括与药品直接接触的包装组件和提供额外保护的二次包装组件。 作为药品包装部件,一方面应满足包装系统的密封要求,为药品提供保护,符合包装的预期使用功能; 另一方面,它应该与药物具有良好的相容性,即不应该引入安全风险。 浸出物或浸出物水平符合安全要求,不会因吸附药物中的活性成分或功能性辅料而影响药物的质量、功效和安全性。 本文主要从两个方面解读兼容性研究的流程和主要内容。
1 兼容性研究过程
参考相关指导原则,包装系统兼容性研究过程主要分为以下六个部分。
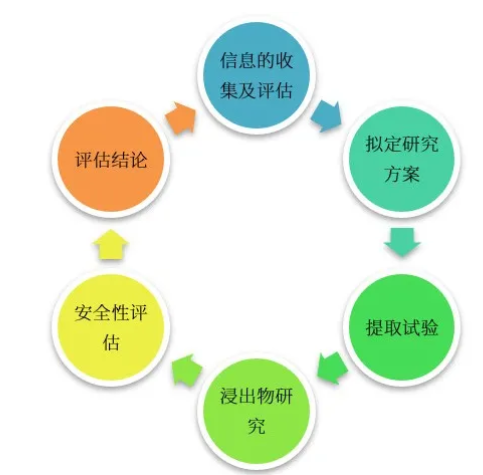
图1 封装系统兼容性研究流程
01信息收集与评估
❏ 确认包装系统各组成部分的法规合规性和质量标准合规性并获取认证材料;
❏ 收集包装系统各部件的配方信息、加工助剂信息、清洗剂和清洗方法等基本信息,以获得添加剂和引入杂质的信息以供研究;
❏ 收集处方组成、关键工艺参数、规格、装量、储存条件、给药途径、给药方法、临床每日最大用量等药品相关信息,以确定提取条件和杂质研究限度。
02 制定研究计划
根据包装系统和药品的特点,合理设计提取试验和/或模拟提取试验。 该计划需要结合USP章节[5]、[6]、[7]、[8]中的相关内容进行设计。 对于具有多种包装的同一规格、材质的品种,应选择比表面积最大的包装系统进行提取试验。 必须针对不同的包装材料或不同的来源进行提取测试。 参照2015年《药包材与药品相容性试验指导原则》的相关要求[9],需要对三批空包装系统进行提取试验和/或模拟提取试验。
03 萃取测试和/或模拟萃取测试
❏ 使用适当的溶剂、药物或模拟药物,选择一定的提取方法和提取条件,在更严格的条件下对包装系统的各组分或整个包装系统进行提取试验和/或模拟提取试验。 对各成分中可萃取的无机物和有机物进行定性和定量研究,利用化学分析进行初步风险评估,预测可能的目标浸出物,并根据研究过程获得已知可萃取物的类型和含量信息。 建立灵敏且专有的分析方法来指导后续的浸出研究;
❏ 安瓿和注射瓶需要首先评估玻璃脱落的风险。 可以参考《ASTM F 1980:2002 无菌医疗器械包装加速老化测试标准指南》[10],取0个月的样品进行加速条件下的腐蚀测试。药品包装材料的相容性,在短期内,获得了与长期条件相当的产品。 同时用氢氧化钠溶液制备阳性对照样品,用扫描电子显微镜(SEM)检测。 同时关注Si、B、Al元素的变化趋势和跳跃点。 跳跃可能表明脱离的风险较高。
04 可浸出物研究
使用所用包装系统对用于商业包装(3 批次)的药品进行了浸出研究(迁移测试)。 对渗滤液检测方法进行充分的方法学研究,确认该检测方法能够特异性、准确、灵敏、稳定地检测渗滤液。 迁移试验一般可与药物稳定性研究一起设计,在稳定性试验的相应时间点检测药物中的浸出物,观察浸出物的变化趋势,并对试验数据进行必要的统计分析和总结。
(注:吸附试验一般与药物稳定性试验同时进行,相容性研究无需审查。)
05 可提取物和/或可浸出物的安全评估
使用列表来评估可提取物和/或可浸出物的安全性。 可以使用PDE法、SCT或QT法、TTC法等计算有机杂质的限量。 您可以使用结构活性数据库和毒理学参考文献(例如互联网)来获取数据。 进行毒性评估; 杂质评估可以参考ICH Q3C[11]、ICH Q3D[12]和ICH M7[13]杂质评估思路。
06 兼容性评估结论
通过提取试验、模拟提取试验、迁移试验结果和安全性评价结果来确定包装系统与药品是否相容以及包装系统是否适用。
2 兼容性研究的主要内容
01提取实验设计
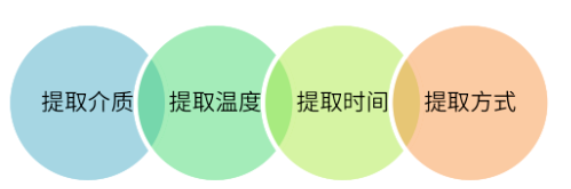
图2 提取实验设计
❏ 提取介质:纯化水、酸性缓冲溶液(pH不应高于药物实际处方,一般选择pH2.5缓冲液)、碱性缓冲溶液(pH不应低于药物实际处方,一般选择pH2.5缓冲液)、碱性缓冲溶液(pH不应低于药物实际处方,一般选择pH2.5缓冲液)。选择pH10.0缓冲液)、复溶药液/液体(若活性成分干扰检测,可选择不含活性成分的空白制剂液)、乙醇水溶液(有机相比例应不低于实际药物处方,一般选择50%乙醇溶液)、正己烷或二氯甲烷(仅适用于气雾剂或特定用途);
❏ 提取条件(温度、时间):一般需要考虑生产过程中可能的加热因素,如冻干过程中最高温度的持续时间、最终灭菌温度和持续时间、最高温度和持续时间。 一般选择40℃/60℃/121℃; 30分钟/60分钟/12小时/24小时。 可根据工艺特点选择试验设备,烘箱、振荡水浴锅或灭菌器;
❏ 提取方法:指药品生产工艺信息以及药品在生产、储存、运输和使用过程中可能面临的最极端条件。 一般采用切割、浸泡、摇动提取或整个装置摇动提取(可采用直立或倒置方法)。 其中,涂层胶塞可根据其涂层功能将整个胶塞浸泡提取,无需将其切成碎片。
02 浸出试验设计
图3 浸出试验设计
测试条件:参考说明书中的储存条件和品种特性,结合ICH指导原则和中国药典稳定性指导原则,制定稳定性设定条件。 一般稳定品种检验条件:
加速度:40℃±2℃,75%±5%或30℃±2℃,65%±5%;
长期:25℃±2℃,60%±5%;
请注意,根据 ICH 指南,半渗透性材料需要低湿度检查条件;
❏ 测试时间:根据产品有效期设计放样点,高于有效期添加放样点,一般按照加速3个月、加速6个月、长期6个月、长期12个月、长期-期限24个月,有效期,超过有效期。 品种规格多,稳定性检验样品多。 所有规格都需要进行测试。 同时要注意不同材质包装系统的摆放方式。 一般玻璃安瓿和半透材料可以平放,注射瓶+胶塞可以直立或倒置。 (如加速3个月、加速6个月、长期6个月、有效期关键点均进行检测。其他常规采样点优先采用倒样方式进行检测,如果无明显变化杂质和元素杂质浸出的风险被认为较低,因此不需要再次测试直立样品。
❏ 调查项目:通过提取试验和模拟提取试验(可浸出预试验)评价有机物和无机物,以确定需要继续进行渗滤液研究的杂质。 其中药品包装材料的相容性,Cd、Pb、As、Hg、Co、V、Ni、Li、Sb、Cu、Si、B、Al等13种元素需要研究。
03 可提取物和浸出物的检测方法
使用ICP-MS、ICP-OES、AAS和其他仪器对元素杂质进行半定量和/或定量分析; 使用GC-MS、LC-MS、HPLC、IC、GC和其他仪器对有机杂质进行半定量和/或定量分析。
04 可萃取物和浸出物检测方法的验证
方法验证参照中国药典导则9101《药品质量标准分析方法验证指导原则》[14]:
✔ 半定量方法需要验证系统适用性和灵敏度;
✔ 定量方法需要验证特异性、灵敏度、线性和范围、精密度、准确度等。
05 可萃取物和浸出物的评价与分析
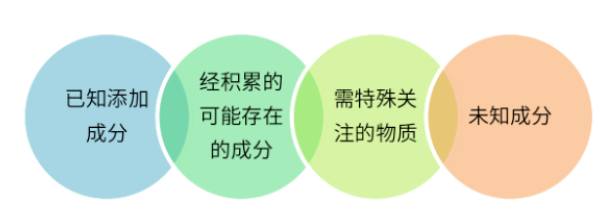
图4 萃取物和浸出物评价分析
❏ 已知添加成分
✔ 通过信息收集获得已知添加成分,通过文献研究获得人体每日允许暴露量(PDE)进行半定量评估;
✔ 如果无法获得PDE值,可以参考ICH Q3C、ICH Q3D、ICH M7等指导原则,通过LD50、TD50、LOEL、NOEL等数据换算得到评价限。 毒性数据可从结构活性数据库(如 DEREK、CCRIS、HSDB、RTECS、CORE、毒理学参考文献)以及互联网上获得。
❏ 可能已积累的成分
由于弹性体成分复杂,变化风险较多,因此有必要积累并建立弹性体可提取物数据库。 评价思路与已知添加成分相同。